这篇笔记的目的是为了学习全基因组测序的步骤和相关软件的简单使用
环境:Windows的Ubuntu子系统
数据:saccharomyces cerevisiae(酿酒酵母)
安装工具
- 进入anaconda网站安装Miniconda,在网站可以找到使用命令行快速安装的链接
1
2
3
4
5
|
conda install -c conda-forge mamba
mamba create -n reseq-pipeline -c bioconda bwa samtools picard gatk4 snpeff sra-tools fastqc
|
1
2
3
4
|
conda activate reseq-pipeline
conda deactivate
|
下载数据
1
2
| mkdir -p genome_resequencing/data
cd genome_resequencing
|
1
2
3
4
| wget -P data/ http://ftp.ensembl.org/pub/release-108/fasta/saccharomyces_cerevisiae/dna/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa.gz
gunzip data/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa.gz
|
1
2
3
4
5
|
wget https://download2.cncb.ac.cn/INSDC5/ERA/1/ERR1727/ERR1727715//ERR1727715.sra
fastq-dump --split-files --gzip -O data/ data/ERR1727715.sra
|
此时得到了两个文件ERR1727715_1.fastq.gz和ERR1727715_2.fastq.gz
处理数据
记录了从原始测序数据 (FastQ) 到变异功能注释 (Annotated VCF) 的标准流程。
流程步骤:
- 使用 FastQC 进行原始数据质量控制。
- 为参考基因组建立 BWA, Samtools, Picard 所需的索引和字典文件。
- 使用 BWA-MEM 将 FastQ 文件比对到参考基因组。
- 使用 Samtools 和 Picard 对 SAM/BAM 文件进行处理(格式转换、排序、去重)。
- 使用 GATK HaplotypeCaller 进行变异检测。
- 使用 SnpEff 对检测到的变异进行功能注释。
使用方法:
- 确保 FastQC, BWA, Samtools, Picard, GATK, SnpEff 已正确安装并配置在系统路径中。
- 将参考基因组 (.fa) 和测序文件 (.fastq.gz) 放入
data/ 目录中。
- 将脚本内容保存为
.sh 文件并赋予执行权限。
脚本安全设置
为了保证脚本的健壮性,在开头进行以下设置:
set -e: 如果任何命令返回非零退出状态,脚本将立即退出。set -u: 如果使用未定义的变量,脚本会报错并退出。set -o pipefail: 如果管道中的任何命令失败,则整个管道的退出状态为失败。
步骤 1: 定义变量
首先,定义脚本中需要用到的所有输入文件、样本信息和工具参数,方便管理和修改。
1
2
3
4
5
6
7
8
9
|
REF="data/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa"
FQ1="data/ERR1727715_1.fastq.gz"
FQ2="data/ERR1727715_2.fastq.gz"
SAMPLE_ID="ERR1727715"
THREADS=4
SNPEFF_DB="R64-1-1.108"
|
步骤 2: 创建输出目录
创建用于存放各个分析步骤结果的目录,使输出文件结构清晰。
1
| mkdir -p qc_reports alignment bam vcf annotation
|
步骤 3: 质量控制 (FastQC)
使用 FastQC 对原始的 FastQ 测序数据进行质量评估,生成的报告有助于了解数据质量。
1
| fastqc "${FQ1}" "${FQ2}" -o qc_reports/
|
步骤 4-6: 建立参考基因组索引
比对和分析前,需要为参考基因组创建多种索引文件,以供不同工具高效调用。脚本会检查索引是否存在,避免重复创建。
- BWA 索引: 用于 BWA-MEM 比对。
- Samtools Fasta 索引: 用于快速访问参考序列的任意区域。
- Picard 序列字典: 主要供 GATK 使用,包含序列名称、长度等信息。
1
| picard CreateSequenceDictionary R="${REF}" O="${REF%.fa}.dict"
|
步骤 7: 基因组比对 (BWA-MEM)
使用 BWA-MEM 算法将双端测序的 reads 比对到参考基因组上,并生成 SAM 格式的比对文件。-R 参数用于添加 Read Group 信息,这对于下游分析(特别是GATK)至关重要。
1
2
| bwa mem -t ${THREADS} -R "@RG\tID:${SAMPLE_ID}\tPL:ILLUMINA\tSM:${SAMPLE_ID}" \
"${REF}" "${FQ1}" "${FQ2}" > "alignment/${SAMPLE_ID}.sam"
|
步骤 8: SAM/BAM 文件处理
对比对生成的 SAM 文件进行一系列处理,为后续的变异检测做准备。
- SAM -> BAM 转换: 将文本格式的 SAM 文件转换为二进制的 BAM 文件,以减小体积和提高处理速度。
1
| samtools view -bS -@ ${THREADS} "alignment/${SAMPLE_ID}.sam" > "alignment/${SAMPLE_ID}.bam"
|
- 排序: 对 BAM 文件按坐标进行排序。
1
| samtools sort -@ ${THREADS} "alignment/${SAMPLE_ID}.bam" -o "bam/${SAMPLE_ID}.sorted.bam"
|
- 标记重复: 使用 Picard MarkDuplicates 标记由 PCR 扩增产生的重复 reads,避免这些重复序列影响变异检测的准确性。
1
2
3
4
| picard MarkDuplicates \
I="bam/${SAMPLE_ID}.sorted.bam" \
O="bam/${SAMPLE_ID}.dedup.bam" \
M="bam/${SAMPLE_ID}.duplicate_metrics.txt"
|
- 创建索引: 为最终处理完成的 BAM 文件创建
.bai 索引,方便快速随机访问。1
| samtools index "bam/${SAMPLE_ID}.dedup.bam"
|
步骤 9: 变异检测 (GATK HaplotypeCaller)
使用 GATK 的 HaplotypeCaller 工具,基于处理好的 BAM 文件进行变异检测(包括 SNP 和 InDel),生成原始的 VCF 文件。
1
2
3
4
| gatk HaplotypeCaller \
-R "${REF}" \
-I "bam/${SAMPLE_ID}.dedup.bam" \
-O "vcf/${SAMPLE_ID}.raw.vcf.gz"
|
步骤 10: 下载 SnpEff 数据库
为了进行功能注释,需要下载对应物种和基因组版本的 SnpEff 数据库。该命令会自动检查数据库是否已存在。
1
| snpEff download -v "${SNPEFF_DB}"
|
步骤 11: 变异功能注释 (SnpEff)
使用 SnpEff 对 VCF 文件中的变异进行功能注释,例如判断变异是发生在基因间区、内含子还是外显子,以及是否导致氨基酸同义/非同义突变等。
1
| snpEff ann -v "${SNPEFF_DB}" "vcf/${SAMPLE_ID}.raw.vcf.gz" > "annotation/${SAMPLE_ID}.annotated.vcf"
|
完整脚本
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
| #!/bin/bash
# ======================================================================================
#
#
#
#
# 1. 使用 FastQC 进行原始数据质量控制。
# 2. 为参考基因组建立 BWA, Samtools, Picard 所需的索引和字典文件。
# 3. 使用 BWA-MEM 将 FastQ 文件比对到参考基因组。
# 4. 使用 Samtools 和 Picard 对 SAM/BAM 文件进行处理(格式转换、排序、去重)。
# 5. 使用 GATK HaplotypeCaller 进行变异检测。
# 6. 使用 SnpEff 对检测到的变异进行功能注释。
#
#
# 1. 确保 FastQC, BWA, Samtools, Picard, GATK, SnpEff 已正确安装并配置在系统路径中。
# 2. 将参考基因组 (.fa) 和测序文件 (.fastq.gz) 放入 `data/` 目录中。
# 3. 将此脚本保存为 `run_full_pipeline.sh`。
# 4. 赋予执行权限: `chmod +x run_full_pipeline.sh`
# 5. 运行脚本: `./run_full_pipeline.sh`
#
# --- 脚本安全设置 ---
# set -e: 如果任何命令返回非零退出状态,脚本将立即退出。
# set -u: 如果使用未定义的变量,脚本会报错并退出。
# set -o pipefail: 如果管道中的任何命令失败,则整个管道的退出状态为失败。
set -euo pipefail
# --- 步骤 1: 定义变量 ---
echo "INFO: [步骤 1/11] 定义输入文件、样本ID和工具参数..."
# 输入文件和样本信息
REF="data/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa"
FQ1="data/ERR1727715_1.fastq.gz"
FQ2="data/ERR1727715_2.fastq.gz"
SAMPLE_ID="ERR1727715"
# 工具参数
THREADS=4 # 使用的线程数
SNPEFF_DB="R64-1-1.108" # SnpEff 使用的数据库版本
echo " - 参考基因组: ${REF}"
echo " - FastQ 1: ${FQ1}"
echo " - FastQ 2: ${FQ2}"
echo " - 样本 ID: ${SAMPLE_ID}"
echo " - SnpEff 数据库: ${SNPEFF_DB}"
echo " - 线程数: ${THREADS}"
echo "--------------------------------------------------"
# --- 步骤 2: 创建输出目录 ---
echo "INFO: [步骤 2/11] 检查并创建所有输出目录..."
mkdir -p qc_reports alignment bam vcf annotation
echo " - 输出目录 'qc_reports', 'alignment', 'bam', 'vcf', 'annotation' 已准备就绪。"
echo "--------------------------------------------------"
# --- 步骤 3: 质量控制 (FastQC) ---
echo "INFO: [步骤 3/11] 使用 FastQC 进行原始数据质量控制..."
fastqc "${FQ1}" "${FQ2}" -o qc_reports/
echo " - FastQC 报告已生成在 'qc_reports/' 目录中。"
echo "--------------------------------------------------"
# --- 步骤 4: 建立参考基因组索引 ---
# BWA index
if [ ! -f "${REF}.bwt" ]; then
echo "INFO: [步骤 4/11] BWA 索引文件不存在,开始为参考基因组建立 BWA 索引..."
bwa index "${REF}"
echo " - BWA 索引创建完成。"
else
echo "INFO: [步骤 4/11] 检测到 BWA 索引已存在,跳过此步骤。"
fi
# Samtools faidx
if [ ! -f "${REF}.fai" ]; then
echo "INFO: [步骤 5/11] Samtools fasta 索引不存在,开始创建..."
samtools faidx "${REF}"
echo " - Samtools fasta 索引创建完成。"
else
echo "INFO: [步骤 5/11] 检测到 Samtools fasta 索引已存在,跳过此步骤。"
fi
# Picard sequence dictionary
REF_DICT="${REF%.fa}.dict"
if [ ! -f "${REF_DICT}" ]; then
echo "INFO: [步骤 6/11] Picard 序列字典文件不存在,开始创建..."
picard CreateSequenceDictionary R="${REF}" O="${REF_DICT}"
echo " - Picard 序列字典创建完成。"
else
echo "INFO: [步骤 6/11] 检测到 Picard 序列字典已存在,跳过此步骤。"
fi
echo "--------------------------------------------------"
# --- 步骤 7: 基因组比对 (BWA-MEM) ---
echo "INFO: [步骤 7/11] 开始执行 BWA-MEM 序列比对..."
bwa mem -t ${THREADS} -R "@RG\tID:${SAMPLE_ID}\tPL:ILLUMINA\tSM:${SAMPLE_ID}" \
"${REF}" "${FQ1}" "${FQ2}" > "alignment/${SAMPLE_ID}.sam"
echo " - 比对完成。输出文件: alignment/${SAMPLE_ID}.sam"
echo "--------------------------------------------------"
# --- 步骤 8: SAM/BAM 文件处理 (Samtools & Picard) ---
# 1. SAM -> BAM 转换
echo "INFO: [步骤 8/11] SAM/BAM 文件处理..."
echo " - (1/4) SAM -> BAM 转换..."
samtools view -bS -@ ${THREADS} "alignment/${SAMPLE_ID}.sam" > "alignment/${SAMPLE_ID}.bam"
# 2. 排序
echo " - (2/4) BAM 文件排序..."
samtools sort -@ ${THREADS} "alignment/${SAMPLE_ID}.bam" -o "bam/${SAMPLE_ID}.sorted.bam"
# 3. 标记重复
echo " - (3/4) 标记重复序列 (Picard MarkDuplicates)..."
picard MarkDuplicates \
I="bam/${SAMPLE_ID}.sorted.bam" \
O="bam/${SAMPLE_ID}.dedup.bam" \
M="bam/${SAMPLE_ID}.duplicate_metrics.txt"
# 4. 创建索引
echo " - (4/4) 为最终 BAM 文件创建索引..."
samtools index "bam/${SAMPLE_ID}.dedup.bam"
echo " - SAM/BAM 文件处理完成。最终文件: bam/${SAMPLE_ID}.dedup.bam"
echo "--------------------------------------------------"
# --- 步骤 9: 变异检测 (GATK HaplotypeCaller) ---
echo "INFO: [步骤 9/11] 使用 GATK HaplotypeCaller 进行变异检测..."
gatk HaplotypeCaller \
-R "${REF}" \
-I "bam/${SAMPLE_ID}.dedup.bam" \
-O "vcf/${SAMPLE_ID}.raw.vcf.gz"
echo " - 变异检测完成。输出文件: vcf/${SAMPLE_ID}.raw.vcf.gz"
echo "--------------------------------------------------"
# --- 步骤 10: 下载 SnpEff 数据库 (如果需要) ---
echo "INFO: [步骤 10/11] 准备 SnpEff 数据库..."
# SnpEff download 命令会自动检查数据库是否存在,如果存在则不会重复下载
snpEff download -v "${SNPEFF_DB}"
echo " - SnpEff 数据库 '${SNPEFF_DB}' 已准备就绪。"
echo "--------------------------------------------------"
# --- 步骤 11: 变异功能注释 (SnpEff) ---
echo "INFO: [步骤 11/11] 使用 SnpEff 进行变异功能注释..."
snpEff ann -v "${SNPEFF_DB}" "vcf/${SAMPLE_ID}.raw.vcf.gz" > "annotation/${SAMPLE_ID}.annotated.vcf"
echo " - 变异注释完成。输出文件: annotation/${SAMPLE_ID}.annotated.vcf"
echo "--------------------------------------------------"
echo "SUCCESS: 所有分析流程已成功完成!"
|
- 这是完事后的文件目录,最终文件是annotation目录下的ERR1727715.annotated.vcf文件

注意事项
R64-1-1.108数据库无法下载
- 由于中国的网络问题,导致无法正常下载snpEff的R64-1-1.108数据库,这里只能手动构建了
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
| #!/bin/bash
# ======================================================================================
#
#
#
#
# 2. 创建 SnpEff 构建数据库所需的目录结构。
# 3. 将基因组和注释文件复制并重命名到指定位置。
# 4. 【智能检查】检查 snpEff.config 文件,如果尚未配置,则自动添加新数据库的条目。
# 5. 【智能检查】检查数据库是否已构建,如果尚未构建,则执行构建命令。
# 6. 使用新建的自定义数据库执行最后的变异注释。
#
# --- 脚本安全设置 ---
# set -e: 如果任何命令失败,脚本将立即退出。
# set -u: 如果使用未定义的变量,脚本会报错并退出。
# set -o pipefail: 如果管道中的任何命令失败,则整个管道的退出状态为失败。
set -euo pipefail
# --- 步骤 1: 定义变量 (请根据您的环境核对这里的路径) ---
echo "INFO: [步骤 1/7] 初始化变量和路径..."
# --- 用户需要确认的变量 ---
# SnpEff 的主目录路径 (请务必确认此路径是正确的)
SNPEFF_DIR="/home/mxrmiss/miniconda3/envs/reseq-pipeline/share/snpeff-5.2-1"
# 您的项目数据目录
DATA_DIR="/home/mxrmiss/genome_resequencing/data"
# 您的 VCF 文件所在目录
VCF_DIR="/home/mxrmiss/genome_resequencing/vcf"
# 您的最终注释文件输出目录
ANNOTATION_DIR="/home/mxrmiss/genome_resequencing/annotation"
# --- 脚本自动化变量 ---
# 参考基因组序列文件
REF_FASTA="${DATA_DIR}/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa"
# 基因注释文件的下载地址 (Ensembl release 108)
GTF_URL="ftp://ftp.ensembl.org/pub/release-108/gtf/saccharomyces_cerevisiae/Saccharomyces_cerevisiae.R64-1-1.108.gtf.gz"
# 下载后的 GTF 压缩文件名
GTF_GZ_FILE="${DATA_DIR}/$(basename ${GTF_URL})"
# 解压后的 GTF 文件名
GTF_FILE="${GTF_GZ_FILE%.gz}"
# 自定义数据库的名称 (可以自己取)
CUSTOM_DB_NAME="sacCer_R64_local"
# SnpEff 配置文件路径
SNPEFF_CONFIG="${SNPEFF_DIR}/snpEff.config"
# 自定义数据库的数据目录
CUSTOM_DB_DATA_DIR="${SNPEFF_DIR}/data/${CUSTOM_DB_NAME}"
# 待注释的原始VCF文件
VCF_IN="${VCF_DIR}/ERR1727715.raw.vcf.gz"
# 注释后的VCF文件
VCF_OUT="${ANNOTATION_DIR}/ERR1727715.annotated.vcf"
echo " - SnpEff 目录: ${SNPEFF_DIR}"
echo " - 自定义数据库名称: ${CUSTOM_DB_NAME}"
echo "--------------------------------------------------"
# --- 步骤 2: 下载并准备基因注释文件 ---
echo "INFO: [步骤 2/7] 准备基因注释文件 (GTF)..."
if [ ! -f "${GTF_FILE}" ]; then
echo " - GTF 文件不存在,开始从 Ensembl 下载..."
wget -P "${DATA_DIR}" "${GTF_URL}"
echo " - 下载完成,正在解压..."
gunzip "${GTF_GZ_FILE}"
echo " - GTF 文件已准备就绪。"
else
echo " - 检测到 GTF 文件已存在,跳过下载。"
fi
echo "--------------------------------------------------"
# --- 步骤 3: 创建 SnpEff 数据库目录并放置文件 ---
echo "INFO: [步骤 3/7] 创建 SnpEff 目录结构并准备文件..."
mkdir -p "${CUSTOM_DB_DATA_DIR}"
# 将文件复制并重命名为 SnpEff 要求的标准名称
cp "${REF_FASTA}" "${CUSTOM_DB_DATA_DIR}/sequences.fa"
cp "${GTF_FILE}" "${CUSTOM_DB_DATA_DIR}/genes.gtf"
echo " - 目录已创建,文件已复制并重命名。"
echo "--------------------------------------------------"
# --- 步骤 4: 自动配置 snpEff.config 文件 ---
echo "INFO: [步骤 4/7] 检查并更新 snpEff.config 文件..."
# 检查数据库条目是否已存在,不存在则添加
if ! grep -q "${CUSTOM_DB_NAME}.genome" "${SNPEFF_CONFIG}"; then
echo " - 在 snpEff.config 中未找到配置,正在添加..."
# 使用 echo 和 >> 操作符在文件末尾追加配置
echo "" >> "${SNPEFF_CONFIG}"
echo "# Custom Yeast genome, R64-1-1 release 108, added by script" >> "${SNPEFF_CONFIG}"
echo "${CUSTOM_DB_NAME}.genome : Saccharomyces_cerevisiae" >> "${SNPEFF_CONFIG}"
echo " - 配置添加成功。"
else
echo " - 检测到配置已存在,跳过此步骤。"
fi
echo "--------------------------------------------------"
# --- 步骤 5: 构建 SnpEff 数据库 ---
echo "INFO: [步骤 5/7] 构建自定义 SnpEff 数据库..."
# 检查构建产物 (snp.bin) 是否存在,如果不存在才执行构建
if [ ! -f "${CUSTOM_DB_DATA_DIR}/snp.bin" ]; then
echo " - 数据库尚未构建,开始执行构建命令..."
snpEff build -gtf22 -v -noCheckCds -noCheckProtein "${CUSTOM_DB_NAME}"
echo " - 数据库构建完成。"
else
echo " - 检测到数据库已构建,跳过此步骤。"
fi
echo "--------------------------------------------------"
# --- 步骤 6: 使用自定义数据库进行注释 ---
echo "INFO: [步骤 6/7] 使用自定义数据库 '${CUSTOM_DB_NAME}' 进行变异注释..."
snpEff ann -v "${CUSTOM_DB_NAME}" "${VCF_IN}" > "${VCF_OUT}"
echo " - 注释完成!"
echo "--------------------------------------------------"
# --- 步骤 7: 完成 ---
echo "SUCCESS: 所有流程已成功完成!"
echo "最终注释文件位于: ${VCF_OUT}"
|
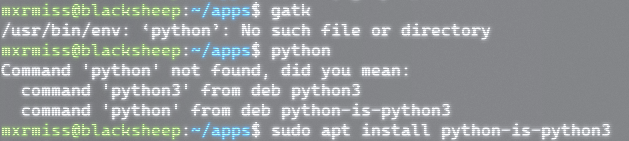
手动安装GATK时出现报错
本文使用conda进行安装测试数据,但也对手动安装GATK出现错误进行了记录
1
| /usr/bin/env: ‘python’: No such file or directory
|
- 原因是Ubuntu会区分python2和python3,所以需要安装一个包,创建一个符号链接,将 /usr/bin/python 指向 /usr/bin/python3
1
| sudo apt install python-is-python3
|