ATAC-seq原理
This article is from: 一文详解ATAC-seq原理+读图:表观遗传的秀儿 - 知乎
ATAC-seq,全称Assay for Transposase-Accessible Chromatin with high throughput sequencing,是2013年由斯坦福大学William J. Greenleaf和Howard Y. Chang实验室开发的用来研究染色质可及性(通常也理解为染色质的开放性)的方法。
ATAC-seq的原理是啥
要理解这项技术的原理,首先要知道染色体/质的结构。真核生物的核DNA并不是裸露的,而是组蛋白与之结合。DNA一圈一圈地缠绕在8个组蛋白上,形成核小体,每个核小体占了约147bp的DNA。一个个核小体构成了串珠样的结构,然后进一步折叠、聚合,并在其他架构蛋白的协助下,形成染色体。这样就能将超长的DNA链,折叠成很小很小的结构,塞进小小的细胞核里。
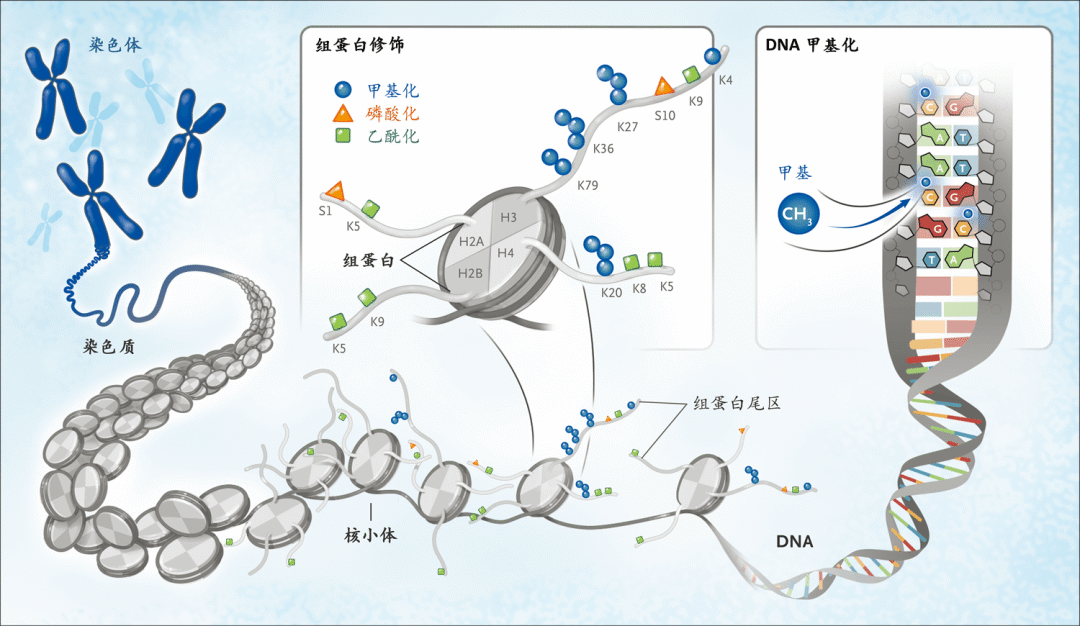
Bates S E . Epigenetic Therapies for Cancer[J]. New England Journal of Medicine, 2020, 383(7):650-663.
但是基因的转录,是需要将DNA的高级结构解开的,但不是需要DNA链全部解开,只需要打开一部分,也就是表达基因的区域解开即可。而这一过程,主要由染色体组蛋白的修饰(尤其是乙酰化)来实现的,这部分打开的染色质,就叫开放染色质(open chromatin)。而染色质一旦打开,就允许一些调控蛋白(比如转录因子)跑过来与之相结合。染色质的这种特性,就叫做染色质的可及性(chromatin accessibility),所以说染色质的可及性反映的就是调控因子与开放染色质结合的状态,与转录调控密切相关。
那我们要如何利用ATAC-seq去找开放的染色质区域呢?
这个技术用到了一个转座酶Tn5:DNA转座是一种由DNA转座酶介导,把DNA序列从染色体的一个区域插入到另外一个区域的现象,类似于“剪切粘贴”。这个过程,也是需要插入位点的染色质是开放的,否则就会被一大坨高级结构给卡住。
既然转座酶Tn5容易结合在开放染色质上,只要人为地将将NGS接头连接到转座酶,携带这些接头的转座酶(如下图带着红色蓝色测序标签的转座酶Tn5)进入细胞核后,在染色质开放区域各种切切切,使染色质断裂并将这些接头插入到开放的染色质区域中,这样我们裂解细胞、破碎DNA后,利用已知序列的测序标签进行NGS测序,就知道哪些区域是开放染色质了。
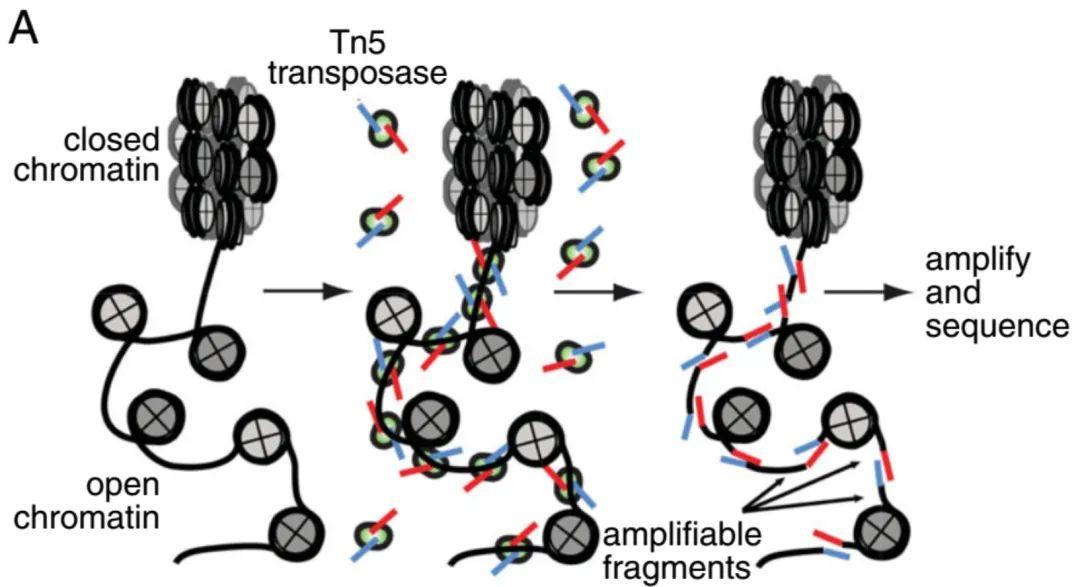
Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015;109:21.29.1-21.29.9.
ATAC-seq和ChIP-seq有啥区别?
ChIP-Seq是实验前明确有一个感兴趣的转录因子,根据目标转录因子设计抗体去做ChIP实验拉DNA,验证感兴趣的转录因子是否与DNA存在相互作用;
而ATAC-Seq没有落脚到具体哪个转录因子,是在全基因组范围内检测染色质的开放程度,可以得到全基因组范围内的蛋白质可能结合的位点信息,用这个技术方法与其他方法结合是想去筛感兴趣的调控因子。
实际上,在全基因组上检测染色质开放程度用DNase-Seq、ATAC-Seq、FAIRE-Seq、MNase-seq都可以,他们又有什么区别呢(如下图)?
MNase-seq和DNase-Seq是用MNase或DNase I内切酶识别开放染色质区域,把切割完的DNA测序,和已知的全基因组序列进行比对,就知道被切掉了哪里,哪里没有被切掉,从而检测出开放的染色质区域。但是实验费时费力,重复性差;
FAIRE-Seq是先进行超声裂解,然后用酚-氯仿富集,不依赖酶和抗体,但弊端就是检测背景高,测序信噪比低,甲醛交联时间不好把握等等;
相比起来,ATAC-seq是用Tn5转座酶,操作起来也更加简单,重复性好,而且最重要的一点是实验只需要很少的细胞/组织量,出来的信号也更加漂亮,所以ATAC-seq目前已经是研究染色质开放性首选的技术方法。
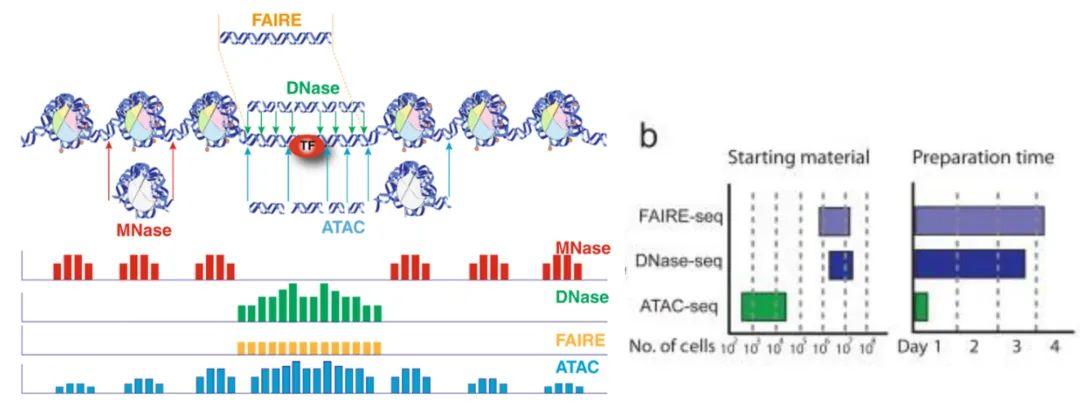
根据这张图我们比较一下这几个技术,ATAC-seq出来的结果,和传统方法出来的结果具有很强的一致性,ATAC-seq也可以剪掉核小体的区域,即作用位点在核小体的两端。中间的大峰是染色质开放区域,两边的小峰为核小体的区域,主要是距离染色质开放区域比较近的地方,随着距离越远,峰值就越来越低了。
常见的染色质开放区有哪些呢?
常见的染色质开放区主要是基因上游的启动子和远端的调控元件比如增强子和沉默子,启动子是靠近转录起始点(TSS)的DNA区域,它包含转录因子的结合位点(transcription factor binding site,TFBS),所以转录因子能够结合在启动子上TFBS,招募RNA聚合酶进而转录基因。增强子一般位于启动子下游或上游1Mb的DNA区域,转录因子与增强子结合,并与启动子区域接触时,能够促进基因的转录。相反,沉默子会减少或抑制基因的表达。
所以说,ATAC-seq可以帮助识别启动子区域、潜在的增强子或沉默子,也就是说,ATAC-seq中的peak,往往是启动子、增强子序列,以及一些反式调控因子结合的位点。
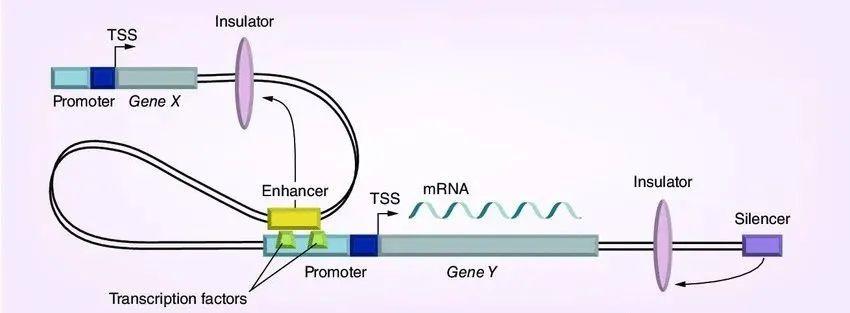
那基因的body不是染色质开放区吗?是的~但是基因body区的染色质开放并不是稳定的,当进行转录时,body区域每往前走一步,开放后进行转录,转录后迅速回复原有状态。
ATAC-seq归根结底能用来干什么?
1、鉴定重要转录因子: 根据原理可以知道,ATAC所捕获染色质开放区一般是正在转录的那部分DNA序列的上下游,得到这些序列我们就可以对富集到的序列结合motif 分析,识别哪种转录因子参与了基因表达调控,最常见的就是去研究转录因子结合的启动子区域(对于抗体质量不好的转录因子,尤其有效)
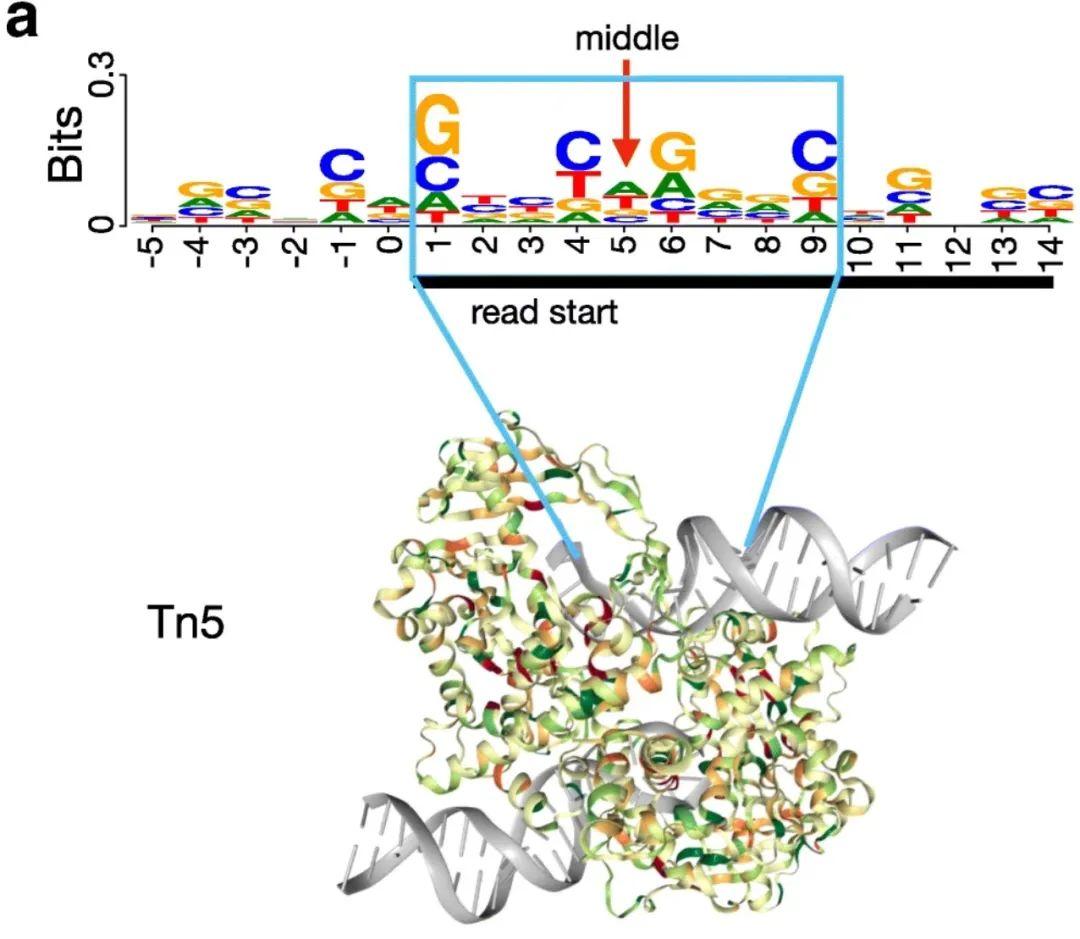
2、生成转录因子结合区域的特征(footprinting): 转录因子结合在DNA上后,它占有的空间阻碍了转座酶Tn5酶切在其他无核小体区域,这样就会留下一个一个小区域,称为足迹(footprint),在这些区域中,reads由高覆盖率峰值突然下降。所以ATAC-seq footprints可以帮助我们查看转录因子在全基因组上结合的状态,主要应用于研究细胞重编程机制,染色质重塑因子,表观修饰对疾病的作用域、T细胞耗竭等等。下面这张图就是已知motif的足迹分析,大概会看到有9个碱基作用的motif
3、生成表观基因组图谱
4、得到在不同组织或不同条件下对应可及性区域。
5、得到核小体位置
最重要的就是数据处理解读这部分了
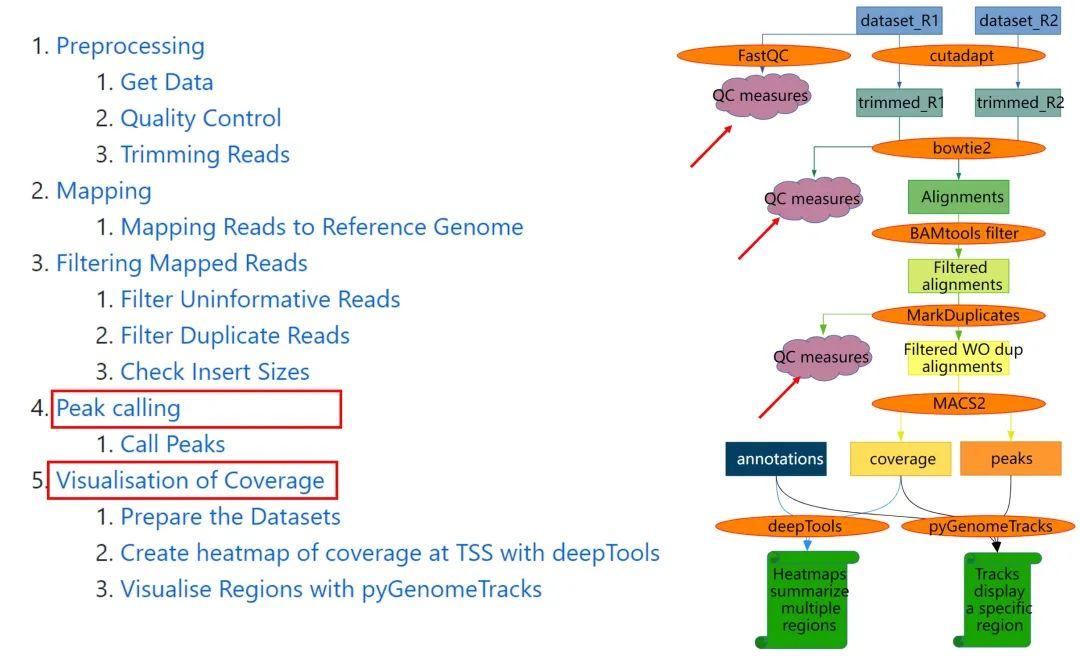
这张图总结了整个数据分析过程,我们重点关注的文章结果图也是主要在Peak calling和Visualisation这两步,如果有小伙伴获得Raw data想自己分析的话,老熊在这里推荐两个教程,个人觉得非常全非常干货:
①https://yiweiniu.github.io/blog/2019/03/ATAC-seq-data-analysis-from-FASTQ-to-peaks/
这两个教程非常细致地讲解了从Raw data到ATAC-seq 的peak数据的分析流程,数据处理这部分就不多赘述了,不过这里老熊要特别强调一下质控这步帮助大家理解:
上图我们可以看到,每一步分析(红色箭头)都涉及到质控,质控对于得到正确的分析结果至关重要:ATAC-seq的质控主要是对文库中的插入标签以及TSS信号进行分析,具体可参照下图理解:
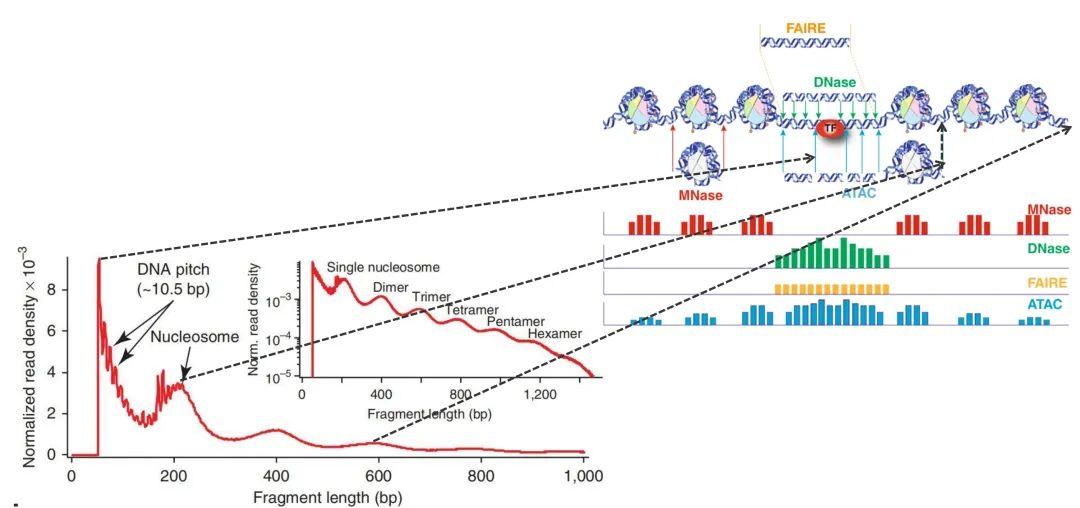
我们可以发现在150bp前出现第一个峰,这个峰值代表的就是切掉了开放的染色质区域,同时伴随着规律的锯齿状小峰,基本是10bp一个小锯齿。在200bp左右处的峰值主要是由于切割核小体导致的,前面我们提到了,一个核小体缠绕的DNA大约在147bp,加上切割的时候不可能那么精确,所以大概在200bp。后续的峰值代表切下来两个、三个核小体以及n倍核小体情况,而且峰值是越来越低的,说明切割到染色质远端的核小体的概率是越来越低的。
结果图怎么看?
下面,就到了大家读文章时都抓耳挠腮的结果图部分,我们重点来看一下Peak callig和Visualisation:
“Peakcalling”
ATAC-seq与ChIP-seq calling出来的peak代表的意义是不同的:
ChIP-seq是用目的蛋白的抗体去拉蛋白,进而把目的蛋白结合的DNA片段也拉下来,然后把 DNA 片段映射到基因组,在基因组上的结合位置就会有DNA 片段堆叠,将这些DNA片段堆叠用柱状图画出来,就会得到所谓的Peak,所以这种情况一般只有一个峰;
而ATAC-seq是靠Tn5转座酶切开染色质开放的DNA位点,Tn5转座酶Tn5在染色体上结合是概率事件,如何判断这个位置的reads足够为一个peak,要用软件MACS统计检测。当转录因子结合DNA时,会阻止Tn5转座酶在该位点上切割,所以会形成一个保护区域,reads无法富集到中间的部分,所以ATAC-seq转录因子结合的区域,一般会有一个山谷般的峰。
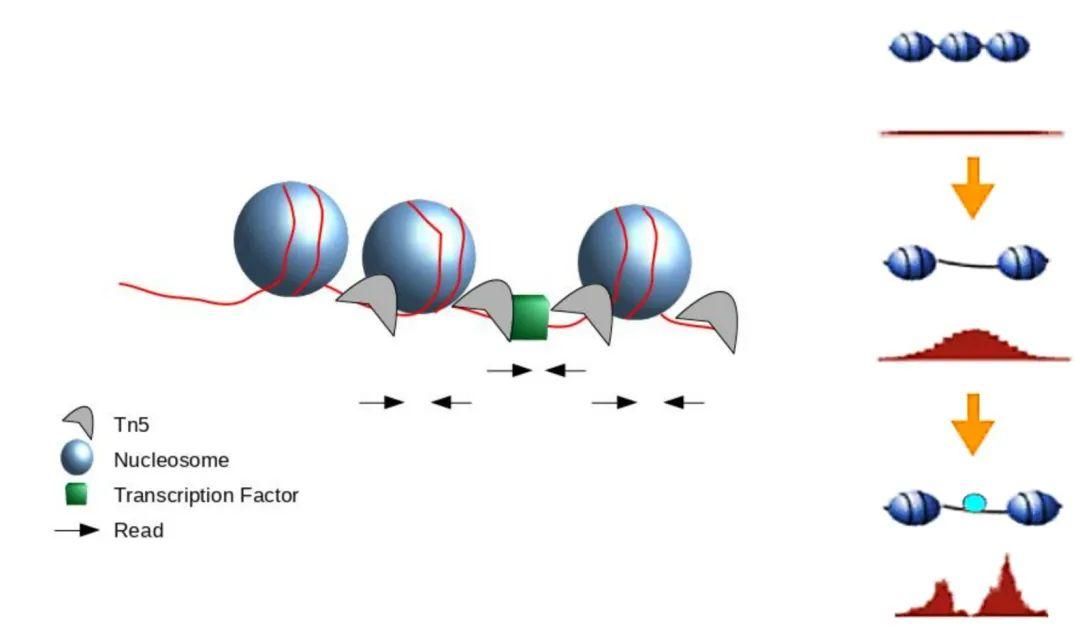
在用MACS软件进行peak calling的时候,建立模型的方式会影响MACS是如何判断peak的标准:
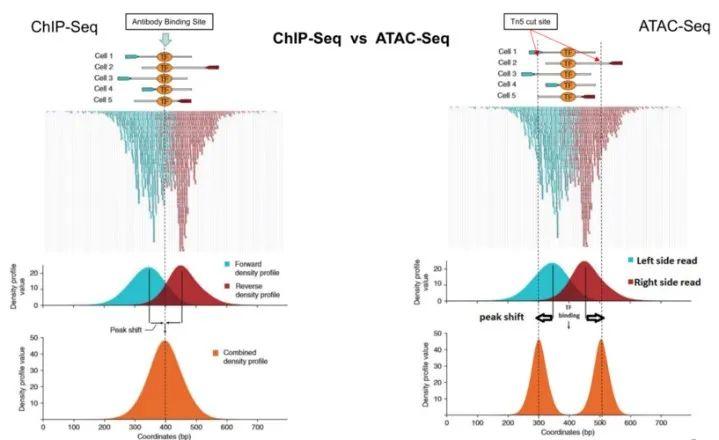
ChIP-seq和ATAC-seq在TF或者Tn5结合区域都会形成一个双峰的reads结合模式,ChIP-seq是由于TF一起沉淀下来的DNA片段一般会大于TF的结合区域,read的位置并不是真实的TF结合的位置,需要向内shift;
而ATAC-seq建模的时候也需要shift,使两个“相邻”的峰shift成一个峰,但是要往两边shift.
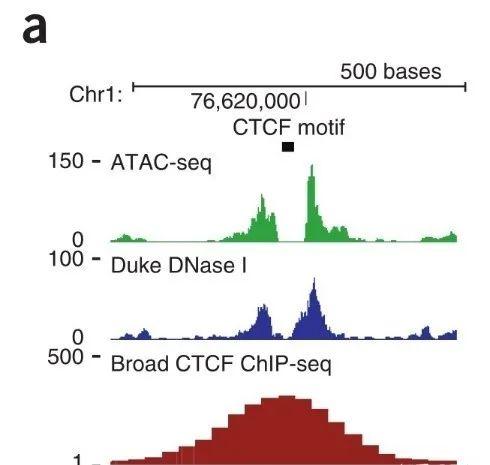
以下图CTCF为例,ChIP-seq的峰就是CTCF的结合区域,中部位置为CTCF的motif,而 ATAC-seq的reads富集在motif两端,横向代表基因组坐标, 纵向代表ATAC-seq的信号强度。
“Visualisation可视化部分”
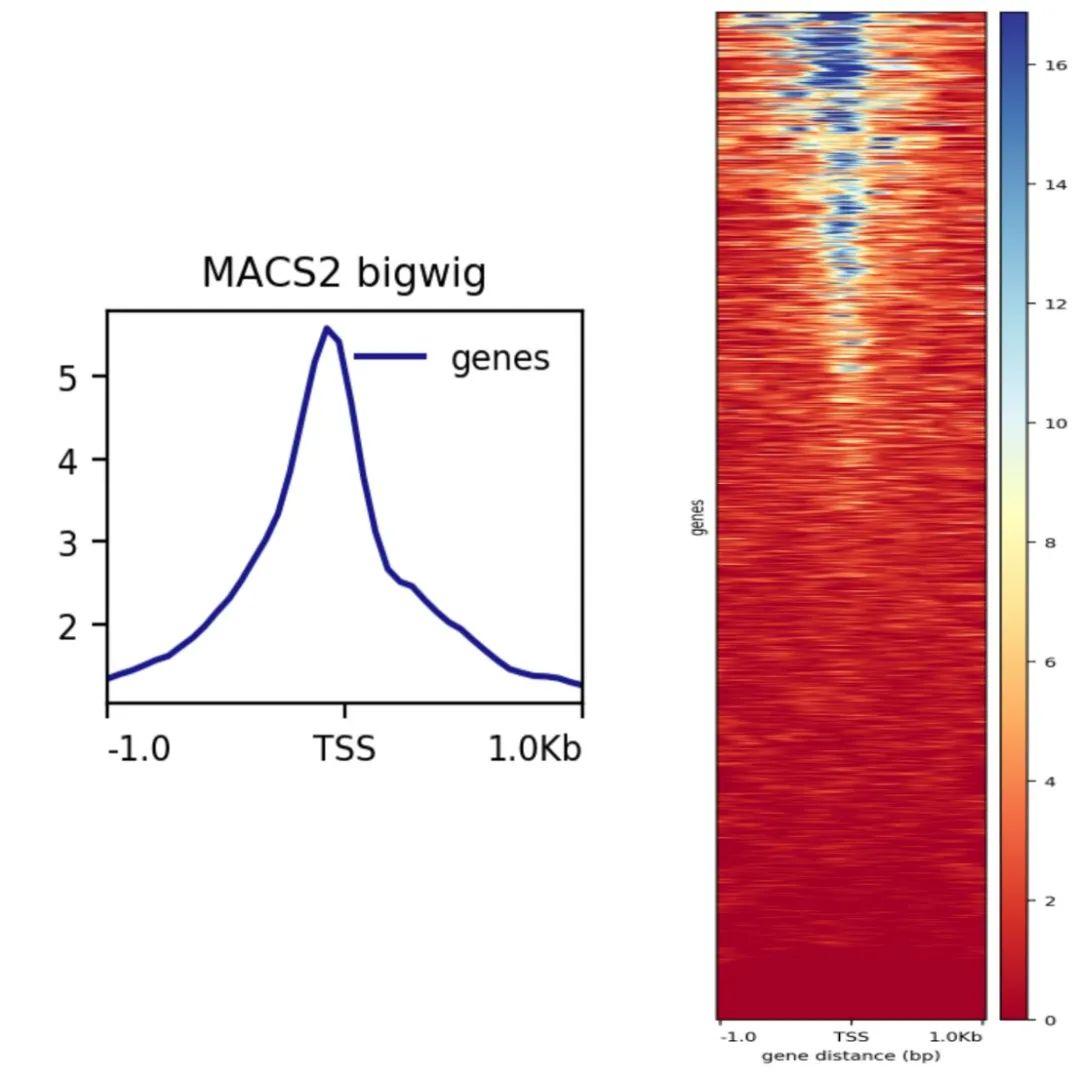
首先我们在文章中经常可以看到以转录起始位点 (TSS) 为中心的峰图和热图(如下图),Each line will be a transcript,通过染色质开放与否识别启动子区域、潜在的增强子或沉默子,因为启动子区域没有明确定义,在基因内部或距离TSS 2.5kb是比较常用的范围。
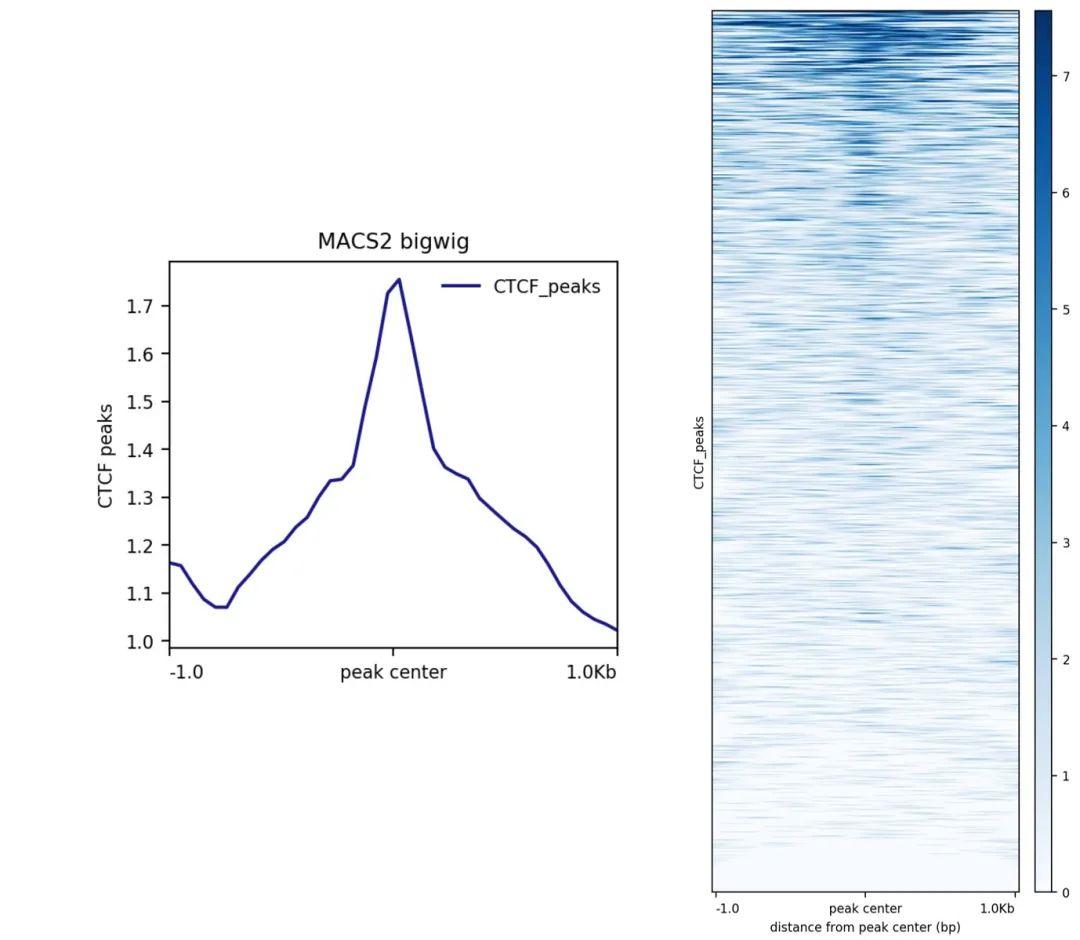
除了以转录起始位点 (TSS) 为中心,也可以特定基因peak为中心作图(如下图):
另外,文章里常可以看到可视化一个特定DNA区域(如下图RAC2基因)
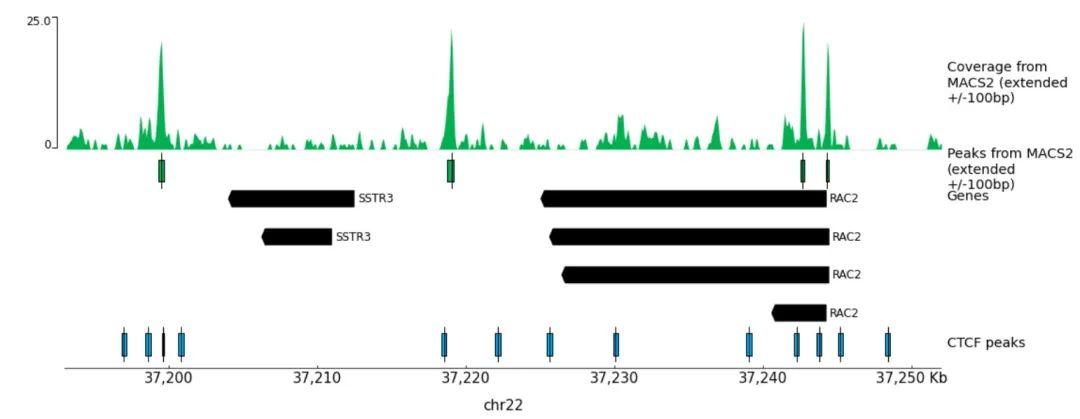
可以看出,在RAC2基因染色质开放区域,对应着CTCF转录因子结合。
做了ATAC-seq可以不做ChIP-seq吗?
答案是No no~~,实际上ATAC-seq在应用中往往要与其他的测序一起联用进行组合分析:
1、ATAC-seq+RNA-seq:
一般,RNA-seq会优先于ATAC-seq先测,得到表达差异基因后,可以通过ATAC-seq来做motif分析,寻找是谁调控了目的基因,然后再进行后续的实验验证。
另一个思路是,看ATAC-seq测到的染色质开放DNA区域,对应的转录本表达量是否也有增加,这样再做RNA-seq可以找到对应的转录本相关基因,对富集到的基因进行功能分析,再结合实验表型验证,从而把一条线串起来:表观调控-表达-功能-表型
2、ATAC-seq+ChIP-seq:
ATAC-seq之后需要做ChIP-seq来做进一步的验证。比如ATAC-seq得到peak后,从motif筛到了转录因子,那么结合ChIP-seq查看转录因子的作用位点,就可以知道是作用在promoter区域呢还是enhancer区域。
除此之外,由于单细胞RNA-seq的普及,目前还出现了一些新兴的scATAC-seq+scRNA-seq,用来检测每个细胞的染色体开放情况。