生信数据格式
生物信息学中常用的数据格式主要用于存储、处理和传输基因组、蛋白质等生物分子数据,以下是对它们的通俗描述:
一、核酸序列相关格式
FASTA 格式
- 用途:存储核酸(DNA/RNA)或蛋白质的序列信息。
- 特点:
- 以
>开头作为序列标识(后面跟名称和注释),例如:>human_gene1 - 接下来的每行是序列字符(A、T、C、G 等),可换行但无其他符号。
- 以
- 例子:
plaintext
1 | |
FASTQ 格式
- 用途:存储高通量测序数据(如 Illumina 测序结果),包含序列和质量分数。
- 特点:
- 每 4 行一组:第 1 行以
@开头的序列标识;第 2 行是序列;第 3 行以+开头(可重复标识);第 4 行是质量分数(每个字符对应序列碱基的测序质量)。
- 每 4 行一组:第 1 行以
- 例子:
plaintext
1 | |
- 质量分数通俗理解:字符对应的数值越高,碱基识别的准确性越高(如
F对应 ASCII 码 70,即质量分数 34,错误率约 1/1000)。
二、基因组注释格式
GTF/GFF3 格式
- 用途:记录基因、外显子、内含子等基因组结构的位置和注释信息。
- 特点:
- 文本格式,每行代表一个注释条目,包含染色体、来源、特征类型(如 gene、exon)、起始 / 结束位置、得分、 Strand(正负链)、相位和属性(如基因名、ID)。
- 例子:
plaintext
1 | |
BED 格式
- 用途:简洁记录基因组区域的位置信息,常用于可视化(如 UCSC Genome Browser)。
- 特点:
- 至少 3 列:染色体、起始位置、结束位置(从 0 开始计数,左闭右开),可扩展列(如名称、得分、链方向等)。
- 例子:
plaintext
1 | |
三、测序比对格式
SAM/BAM 格式
- 用途:存储测序 reads 与参考基因组的比对结果。
- 特点:
- SAM是文本格式,BAM是其二进制压缩版(更省空间,处理速度快)。
- 每行代表一个 read 的比对信息,包含序列名称、比对标志(如是否配对、是否反向)、染色体、位置、匹配质量、CIGAR 字符串(描述比对方式,如 M = 匹配,I = 插入,D = 删除)等。
- 例子(SAM 格式部分列):
plaintext
1 | |
VCF 格式
- 用途:存储基因组变异信息(如 SNP、Indel)。
- 特点:
- 以
#开头为注释行,后续每行代表一个变异位点,包含染色体、位置、参考碱基、变异碱基、质量值、过滤状态、注释信息等。
- 以
- 例子:
plaintext
1 | |
- 通俗解释:chr1 染色体 1500 位置,参考碱基 G 变异为 A,质量值 100,通过过滤,群体中 A 等位基因频率 0.5。
四、蛋白质相关格式
PDB 格式
- 用途:存储蛋白质三维结构数据(原子坐标、化学键等)。
- 特点:
- 文本格式,包含多个部分(如 HEADER、ATOM、TER、END),记录每个原子的类型、位置、所属氨基酸等。
- 例子(部分行):
plaintext
1 | |
FASTA 格式(蛋白质)
- 用途:同核酸 FASTA,只是序列字符为氨基酸(如 A = 丙氨酸,P = 脯氨酸)。
- 例子:
plaintext
1 | |
五、 Plink文件格式
Plink常用的文件格式有两套:map/ped和bim/fam/bed,其中map/ped两种文件格式是互相关联的,通常是一起使用的。两组文件均没有列名,且每一列表示的意思是一定的,格式之间可以相互转换。推荐使用bim/fam/bed这种格式,读取速度快。
map/ped
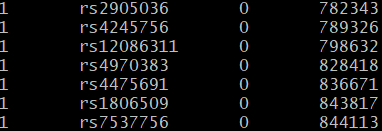
map文件
- map文件(variant information text file)主要记录变异位置信息,由四列构成(map文件没有列名,无Header信息),其包括染色体名称, 所在的染色体和所在染色体的坐标。
(1)第一列:染色体编号;
(2)第二列:SNP位点名称;
(3)第三列:染色体的摩尔位置,即遗传距离。遗传距离通常没有,可以使用0代替,也可以使用-9,-9在Plink中代表缺失;
(4)第四列:SNP物理坐标;
ped文件
- ped文件也是一个无表头的文本文件,它包含了研究对象的系谱和基因型信息。在ped文件中,每行都代表了一个研究对象,通常包括个体ID, 系谱信息, 表型和SNP的分型信息。
(1)第一列: Family ID # 家系编号,如果没有, 可以用个体ID代替;
(2)第二列: Individual ID # 个体ID编号,Family ID和Individual ID连起来必须能够唯一表示每个样本;
(3)第三列: Paternal ID # 父本编号;
(4)第四列: Maternal ID # 母本编号;
(5)第五列: 性别 # Sex(1=male; 2=female; other=unknown), 其他数字表示unknown,通常用0表示;
(6)第六列: Phenotype (0=unknown; 1=unaffected; 2=affected) # 表型数据, 其中表型可以是离散型的(比如质量性状),也可以是连续型的(比如数量性状),plink会自动识别对应的类型。通过以上6个必须的字段,可以完整的映射到某一性状的家系图上。如果未知, 用0表示,也可以使用-9,-9在Plink中代表缺失;
(7)第七列以后:为SNP分型数据, 每个snp位点的基因型需要两列来表示,分别表示major allel和minor allel。在表示基因型时,可以使用A,C,G,T字母的形式,也可以采用1,2数字编码的形式。默认情况下,用0来表示基因型的缺失。
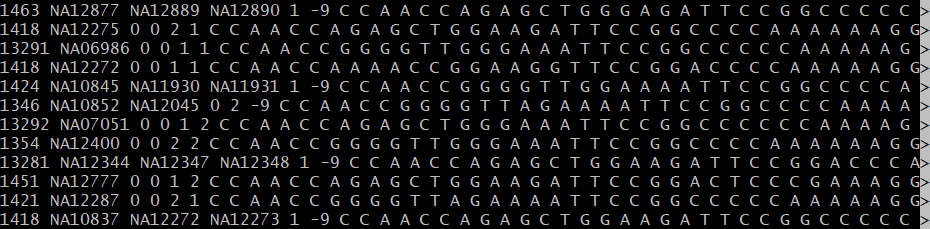
bim/fam/bed
bim文件
BIM文件是一个文本文件,它包含了一组SNP(单核苷酸多态性)位点的位置和名称信息,与MAP文件格式类似。在BIM文件中,每行都代表了一个SNP位点,通常包括以下信息:
(1)第一列:染色体编号(常用整数标记,如22表示第22条染色体,性染色体和线粒体染色体用”X”/”Y”/”XY”/”MT”表示,而0代表染色体信息缺失);
(2)第二列:变异标识符,这个就相当与每一个遗传变异的编号,常见的SNP可以采用以“rs”开头的编号;
(3)第三列:每个遗传变异在基因组上的位置,用摩尔根或者厘摩尔根表示;
(4)第四列:碱基对的坐标;
(5)第五列:等位基因1(A1),通常是次要等位基因(minor allele);
(6)第六列:等位基因2(A2),通常是主要等位基因(major allele)。

fam文件
fam文件也是一个文本文件,它包含了研究对象的个体信息,与ped文件格式类似。在fam文件中,每行都代表了一个研究对象,这些信息通常用制表符分隔,通常包括以下信息:
(1)第一列:家系编号(“FID”);
(2)第二列:个体编号(“IID”; 不能是”0”);
(3)第三列:父系编号 (“0”表示父系信息缺失);
(4)第四列:母系编号(“0”表示母系信息缺失);
(5)第五列:性别编号(1= 男, 2=女, 0=性别未知);
(6)第六列:表型值 (1=对照,2=病例, -9/0/表示表型缺失)。

六、其他常用格式
BEDGRAPH/WIG 格式
- 用途:存储基因组信号强度(如测序覆盖度、甲基化水平)。
- 特点:
- BEDGRAPH:3-4 列,染色体、起始、结束、信号值;
- WIG:更灵活,可记录固定步长的信号值。
GFF 格式(通用特征格式)
- 用途:类似 GTF,用于描述各种生物特征,格式更通用。
Vienna 格式
- 用途:存储 RNA 二级结构(如碱基配对信息),例如:
ACGUACGU.(..)..,点和括号表示配对关系。
总结
这些格式各有侧重:
FASTA/FASTQ是基础序列格式,后者带质量值;
SAM/BAM用于比对分析,BAM 更高效;
GTF/GFF/VCF是注释和变异的 “字典”;
PDB则是蛋白质结构的 “三维蓝图”。
实际分析中,常需根据工具(如 BWA、GATK)和场景选择或转换格式。