BLAST+使用
本文主要介绍在Linux环境下blast+的安装以及使用
1. 安装
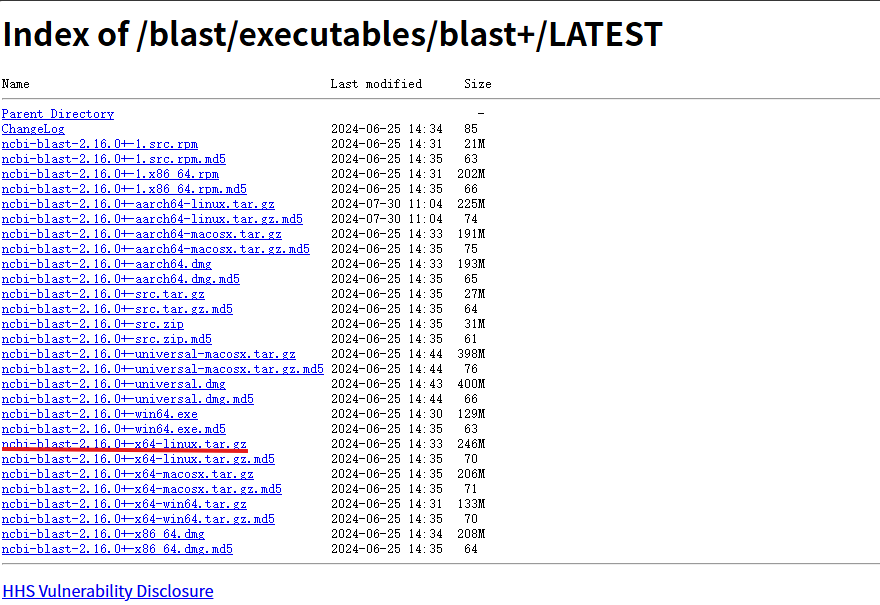
- 进入这个网址https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
- 选择合适的Linux版本,然后复制链接
- 在Linux中拉取安装包
1 | |
- 重命名为blast
1 | |
- 添加环境变量到~/.bashrc,使用vim在该文件的末尾添加代码:
1 | |
- 此时安装完成,但使用命令blastn -help时会报错(缺乏依赖):
1 | |
- 使用命令安装包
1 | |
2. 使用
2.1 安装数据库
- 创建本地数据库目录
1 | |
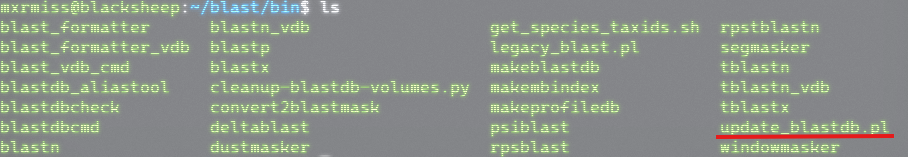
- 进入blast的bin目录, 我们会看到一个perl文件:
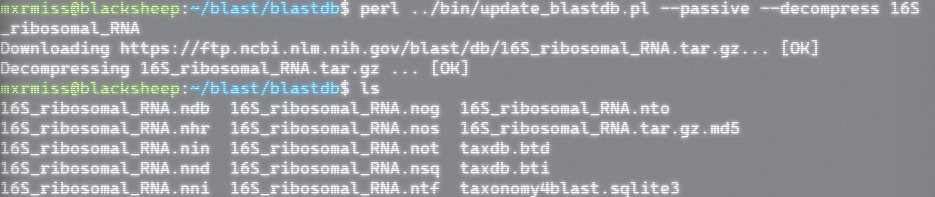
- 进入上面的数据库目录中,执行命令(运行需要时间,需要等待一会儿):
1 | |
- 该命令的目的是使用脚本下载数据库,运行改脚本还可以更新本地的数据库,结果:
- 例子:从本地数据库中搜索NR_025000序列,并以fasta格式保存到16S_query.fa中(注意16S_ribosomal_RNA的文件位置)
1 | |
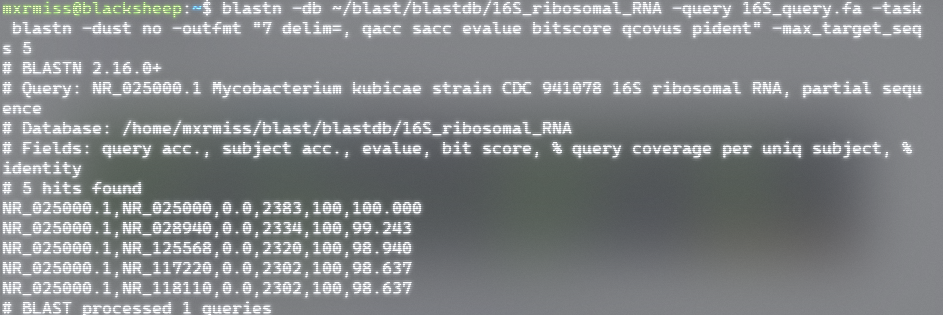
- 用16S_query.fa中的nr_025000序列和本地数据库里的序列进行对比,输出最前面5条结果
1 | |
2.2 案例
- 数据库中包含以下内容

创建数据库
- github上面下载好案例后,创建数据库:
1 | |
-in 是已存在的fasta文件
-dbtype 是数据库类型
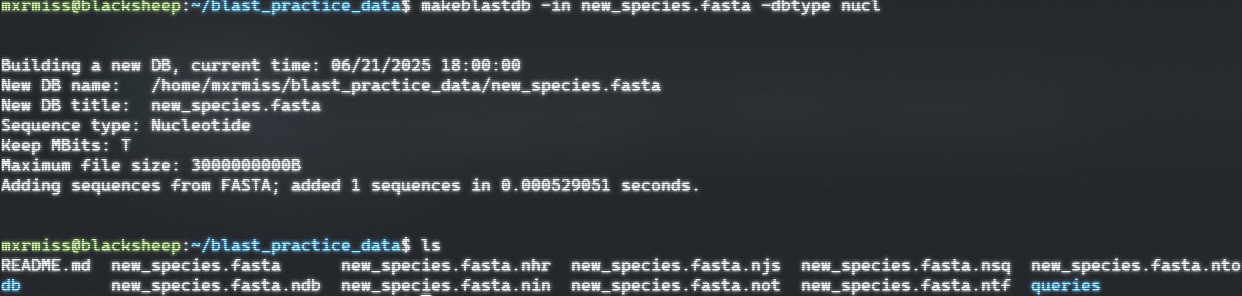
-out 是所创建的数据库名称,如果不指定,默认以当前文件夹为数据库所在地,并且数据库名称默认为-in 参数后面的fasta文件名称
注意:-out 后面的db这是一个文件夹,真正的数据库是new_species
- 不指定 -out 参数创建数据库如下图所示:
blastn查询对比
mismatch.fasta
- 以queries/mismatch.fasta为查询序列进行查询, out1为输出文件, 这里使用了 ‘>’ , 所以可以不使用blastn的 -out 参数
1 | |
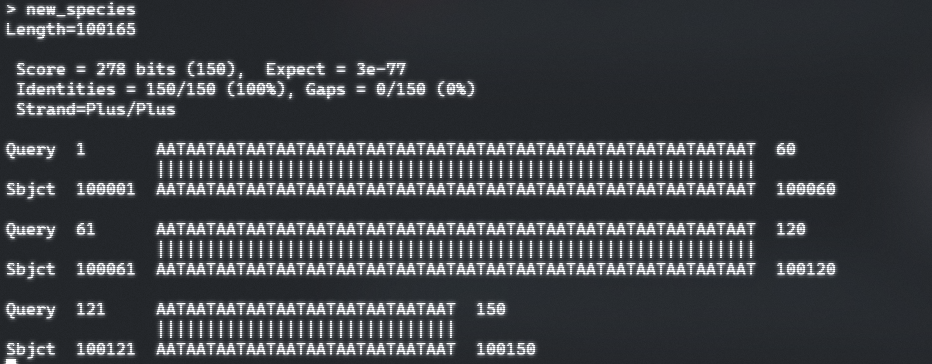
- out1的主要内容是
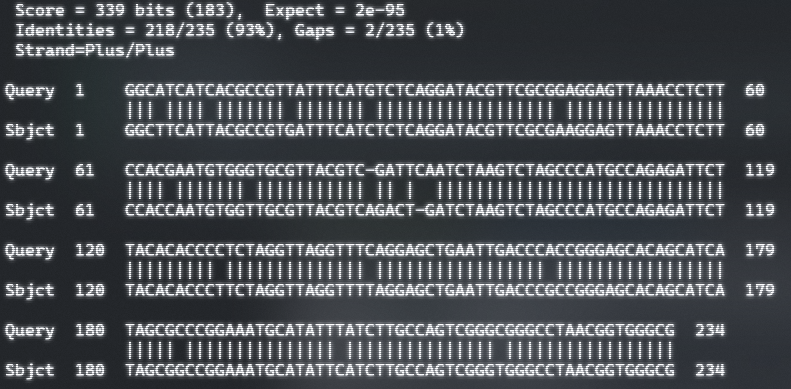
Score是对比得到的分数,这个结果并非最重要的结果
Expect是期望,是最重要的结果,当≤0.01的时候认为结果是显著的,即搜索结果的误差是很小的,但使用不同数据库以及算法的时候的**E值**都需要进行调整
Identities是序列对比的一致性
Gaps是错配(缺少或增加)的片段,用 ‘—‘ 表示
Strand是序列的方向,若查询序列(Query)或参考序列(Sbjct)是正向的,即序列方向是5’->3’,Strand为Plus,若反向的,即为互补链,Strand为Minus
short.fasta
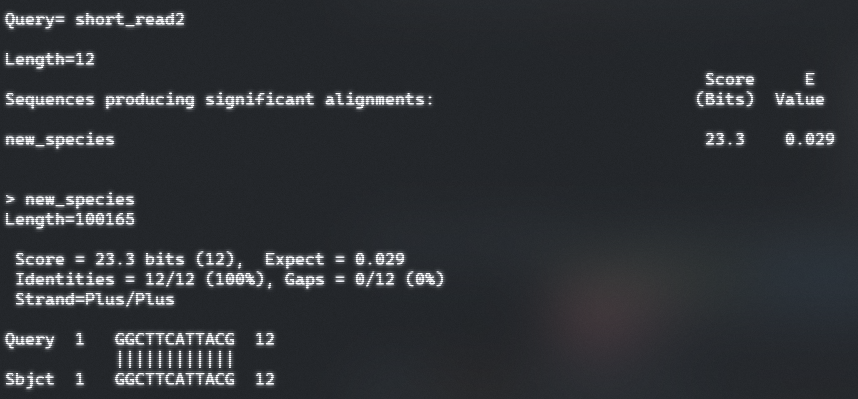
- 以queries/short.fasta为查询序列进行查询, out2为输出文件
1 | |

- 发现结果中并未查询到同源序列,此时使用grep命令让序列和数据库进行匹配会发现数据库中包含了我们要查询的序列,这是因为blast中默认查询序列长度大于我们所搜索的序列长度,

- 使用参数
-word_size可以指定搜索序列长度:
1 | |
- 此时out2文件中出现了两个结果,下面为E值显著的结果:
compliment.fasta
- 下面的例子可以看到Strand中显示两条相反的序列
1 | |
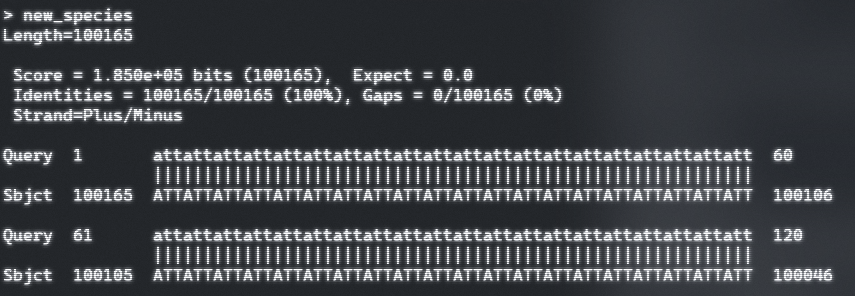
- Blast+默认开启序列互补匹配,如果使用参数
-strand=plus, 我们会发现没有匹配的结果
1 | |
gaps.fasta
- 有时我们发现匹配的结果中会有许多空位片段,如下:
1 | |
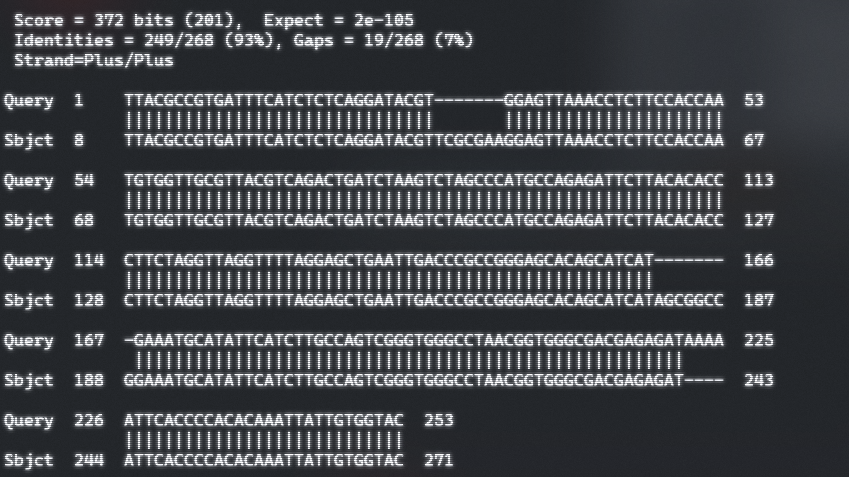
- 为了尽量不匹配到空位,可以使用参数
-gapopen和-gapextend
**
gapopen**:控制是否开启空位(影响空位数量),空格罚分**
gapextend**:控制空位延伸的 “成本”(影响空位长度)
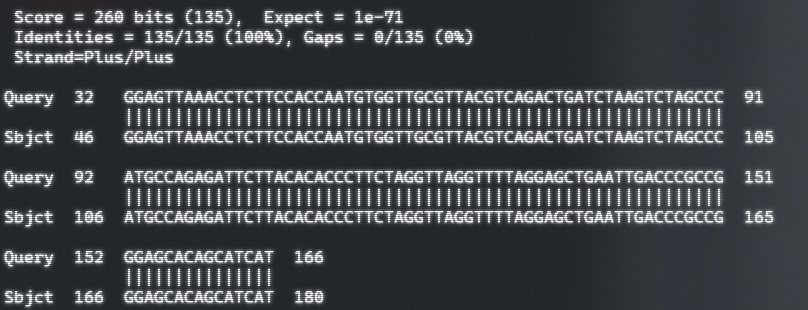
- 如下所示, 空格罚分为100,空格允许最大长度为2,可以发现查询的序列没有空格
1 | |
repeat.fasta
- blast会自动过滤掉一些重复序列,因为这些序列可能在绝大多数生物中都存在,如下所示:
1 | |
- 有时候这些被过滤掉的序列很有用,所以我们可以手动关闭系统的过滤,使用参数
-soft_masking false如下:
1 | |
3. 其他参数
-query: 输入文件路径及文件名;
-out:输出文件路径及文件名;
-db:数据库路径及数据库名;
-task: 共五个程序选择’blastn’ ‘blastn-short’ ‘dcmegablast’ ‘megablast’ ‘rmblastn’ ,默认megablast;
具体区别如下:
blastn 完全匹配的传统blastn;
blastn-short 优化查询:短于50个碱基;
megablast 查找十分相似的序列(如物种内部或相关的物种间);
dc-megablast 查找亲缘关系比较远的序列(如物种间);
rmblastn 兼容了RepeatMasker。
-evalue:设置输出结果的e-value值,一般1e-5;
-num_threads:线程数,笔记本不要设大了,2就够了;
-num_alignments:输出数据库中能与Query比对上的的序列数目,与max_target_seqs不能同时使用;
-max_target_seqs:最多允许比对到数据库中的序列数目,参数仅适用于outfmt >4;
-perc_identity :比对的最低相似度;
-max_hsps:由于不对时一条序列比对成多段,如果只想输出其中的几段,就设定相应的数目,与-num_alignments不能同时使用;
-outfmt:输出文件格式,总共有15种格式,一般设置为6。6是tabular格式对应BLAST的m8格式;
0 = pairwise,
1 = query-anchored showing identities,
2 = query-anchored no identities,
3 = flat query-anchored, show identities,
4 = flat query-anchored, no identities,
5 = XML Blast output,
6 = tabular,
7 = tabular with comment lines,
8 = Text ASN.1,
9 = Binary ASN.1,
10 = Comma-separated values,
11 = BLAST archive format (ASN.1),
12 = JSON Seqalign output,
13 = JSON Blast output,
14 = XML2 Blast output。
此外还能自定义输出格式主要针对上述的 6, 7, and 10三种格式。
outfmt 6输出12列对应的含义:
Query id:查询序列ID标识;
Subject id:比对上的目标序列ID标识;
% identity:序列比对的一致性百分比;
alignment length:符合比对的比对区域的长度;
mismatches:比对区域的错配数;
gap openings:比对区域的gap数目;
q. start:比对区域在查询序列(Query id)上的起始位点;
q. end:比对区域在查询序列(Query id)上的终止位点;
s. start:比对区域在目标序列(Subject id)上的起始位点;
s. end:比对区域在目标序列(Subject id)上的终止位点;
e-value:比对结果的期望值,解释是大概多少次随即比对才能出现一次这个score,Evalue越小,表明这种情况,从概率上越不可能发生,但是现在发生了,所以这个比对具有很重要的意义;
bit score:比对结果的bit score值。bit score值表示对齐的好坏;分数越高,对齐效果越好。一般来说,这个分数是根据一个公式计算出来的,这个公式考虑了相似或相同残基的排列,以及为了排列序列而引入的任何gaps。